
The Enduring Enigma of the FCS
The furin cleavage site should have settled the question of an artificial origin long ago. Certain zoology professors should be made to do a basic statistics course.

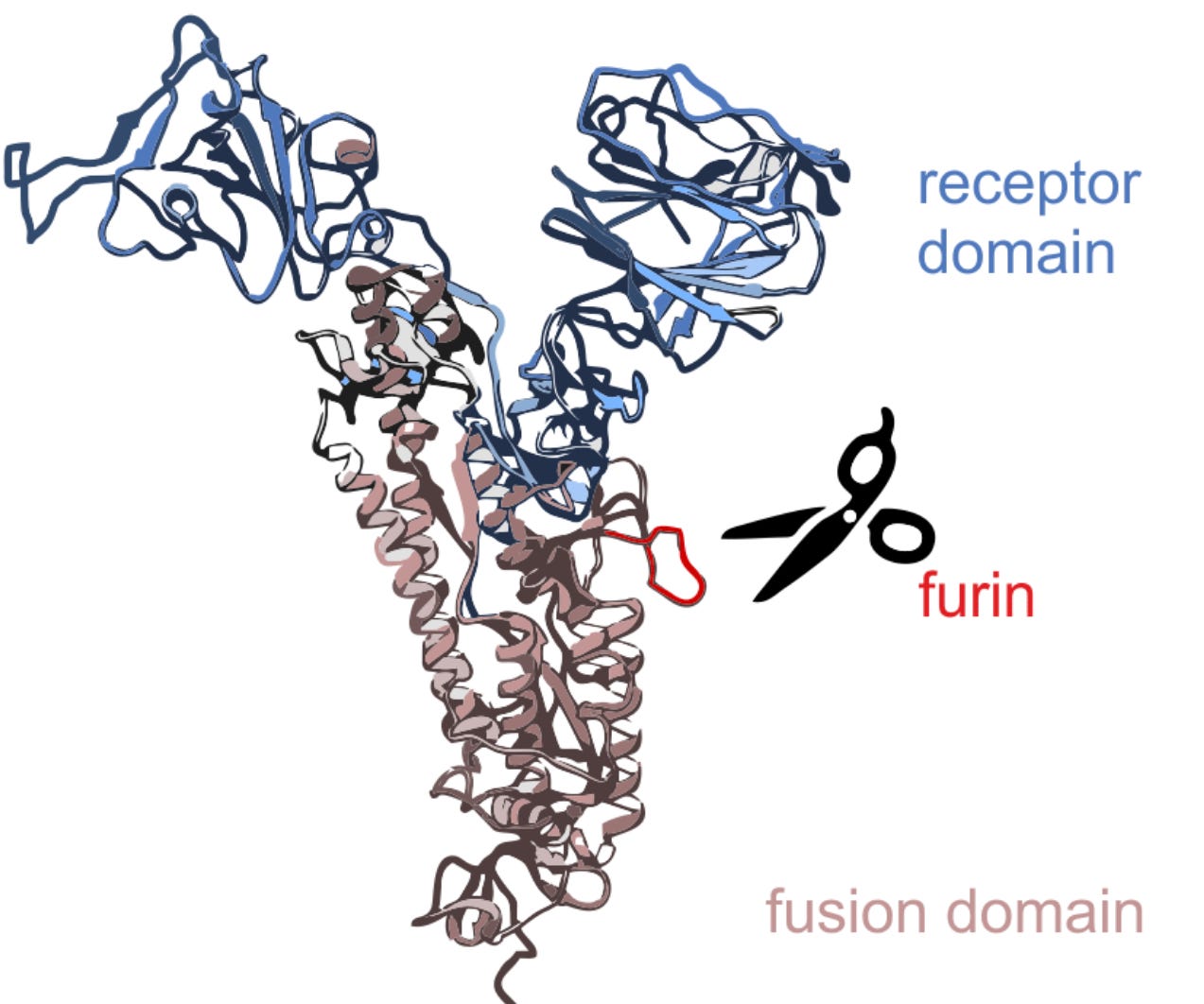
From the start, the Furin Cleavage Site has attracted more attention than any other genomic feature as pointing to an artificial origin. It isn’t simply that all of SARS-CoV-2’s close relatives (even the fakes) lack an FCS motif, it’s also that it occurs as an insert. It didn’t evolve one mutation at a time, it happened in an instant - RNA from an unknown foreign source somehow got injected due to an error during replication. Although such a phenomenon isn’t regarded as impossible, it is exceedingly rare. Even more improbable that the RNA randomly acquired would form a valid cleavage motif, and in the precise location the S1/S2 junction is cleaved. If a detailed calculation multiplying these minuscule probabilities had been made, the debate over an artificial origin should have ended in February, 2020.
Yet it continues. “That’s not how I would have designed an FCS - it’s sub-optimal” they say, “other coronavirus families have an FCS”, “the S1|S2 junction is a hotspot for inserts”, “we don’t know what else may be out there” etc. This shouldn’t be difficult to counter - but for an uneven balance of academic credentials. A cabal of former Oxford Zoology Department professors (Holmes/Worobey/Rambaut/Robinson et al) refuse to engage in serious debate, but recite these same talking points ad nauseum to any media still interested. It perhaps doesn’t help that lab-leakers indulge in speculating on artificial sources it may have come from e.g. “a Moderna patent”, “the human Enac sequence” etc. This shifts the debate from the infinitesimally small chance of it ever having happened, to having to defend a specific source. In an artificial origin, the FCS needn’t match anything, it could easily have been derived from undocumented experiments.
Still, knowing the pitfalls, I’m going to weigh in again anyway…
CpG depletion
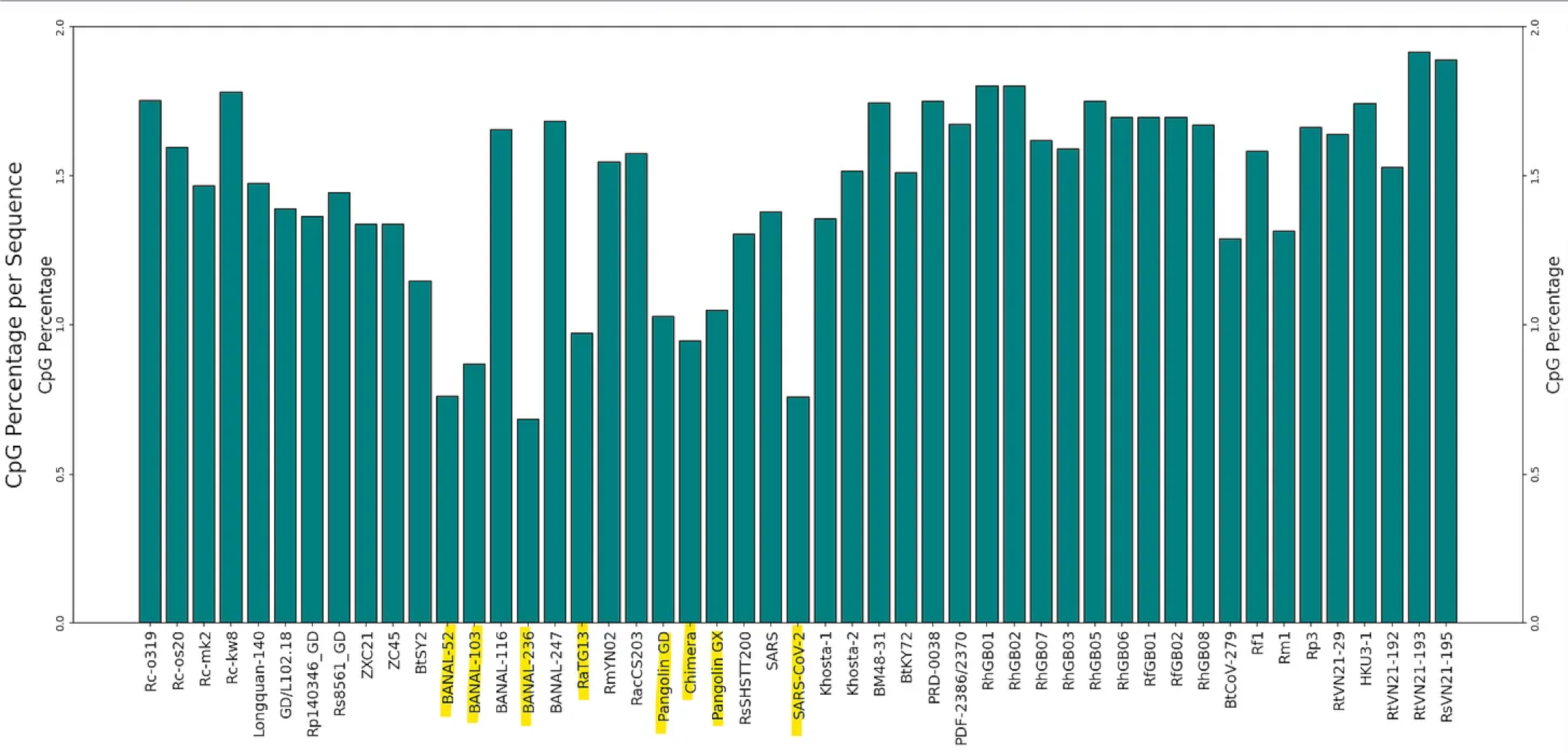
SARS-CoV-2 - and particularly the spike protein is extremely depleted in CpG dinucleotides (all this means as a C followed by a G in the nucleotide sequence). This has been hypothesized to be due to the virus evolving to evade the host immune system, by taking on the hosts own nucleotide bias (human and bat genomes are also CpG depleted). To an extent this is true of all coronaviruses, CG is usually in a range 1.5-3% of all nucleotide pairs, whereas if random, we’d expect 6.25%.
But SARS-CoV-2 is exceptional in that:
the depletion is particularly pronounced in spike (at only ~0.7%). There’s no obvious natural explanation for this. Host factors that seek out CpG don’t know which region of viral RNA corresponds to which gene/protein.
this depletion is also evident in the 3 Banal sequences closest to SARS-CoV-2, RaTG13 and pangolin CoVs, but not in most other bat CoVs. The Banal sequences have an extended region straddling the RBD, where there are no CG at all.
There are many potential causes of CpG depletion in a lab environment. Cytosine deamination causes C→T mutations, so CG becomes TG. This can result from heating (e.g. during PCR), sequencing, irradiation, long-term storage, UV light, exposure to enzymes and other reagents.
But if SARS-CoV-2 spike is depleted in CpG overall, why does the FCS have three CG pairs in such a short segment (it is coded CCT-CGG-CGG-GCA-CGT)? The codon CGG is rare in coronaviruses making up ~3% of Arginine codons, or ~1 in 1000 of all codons. This suggests a source other than a coronavirus. Humans have a higher preference for CGG codons (~20% of Arg or ~6 in 1000 of all codons), but the human genome is also overall CpG depleted (~1%), as are those of bats. The chance of two CGG codons in a row is still only 0.0035% (if random).

So, what might be the source?
If you read my previous article, you’ll know about my woes FOI-ing documents from CSIRO. Almost all documents relating to SARS have disappeared. But they did manage to dig up some documents relating to other projects. I had requested all correspondence from Zhang Huanjun, the WIV intern who was running gain of function experiments in CSIRO’s BSL-4 lab. As a side project, he was sequencing a bat herpesvirus, and exchanging sequence data with CSIRO staff via email.
Not expecting much, I searched anyway for CGGCGG. To my surprise it was everywhere, over 1000 instances, and extending up to 10 nucleotides from the FCS. The explanation is that unlike coronaviruses, this herpesvirus is CpG enriched (~9% of nucleotide pairs). This WIV/CSIRO work resulted in a paper, and a complete herpesvirus genome (>200k bases) posted to GenBank. Blasting it results in longer FCS matches up to 12nt.
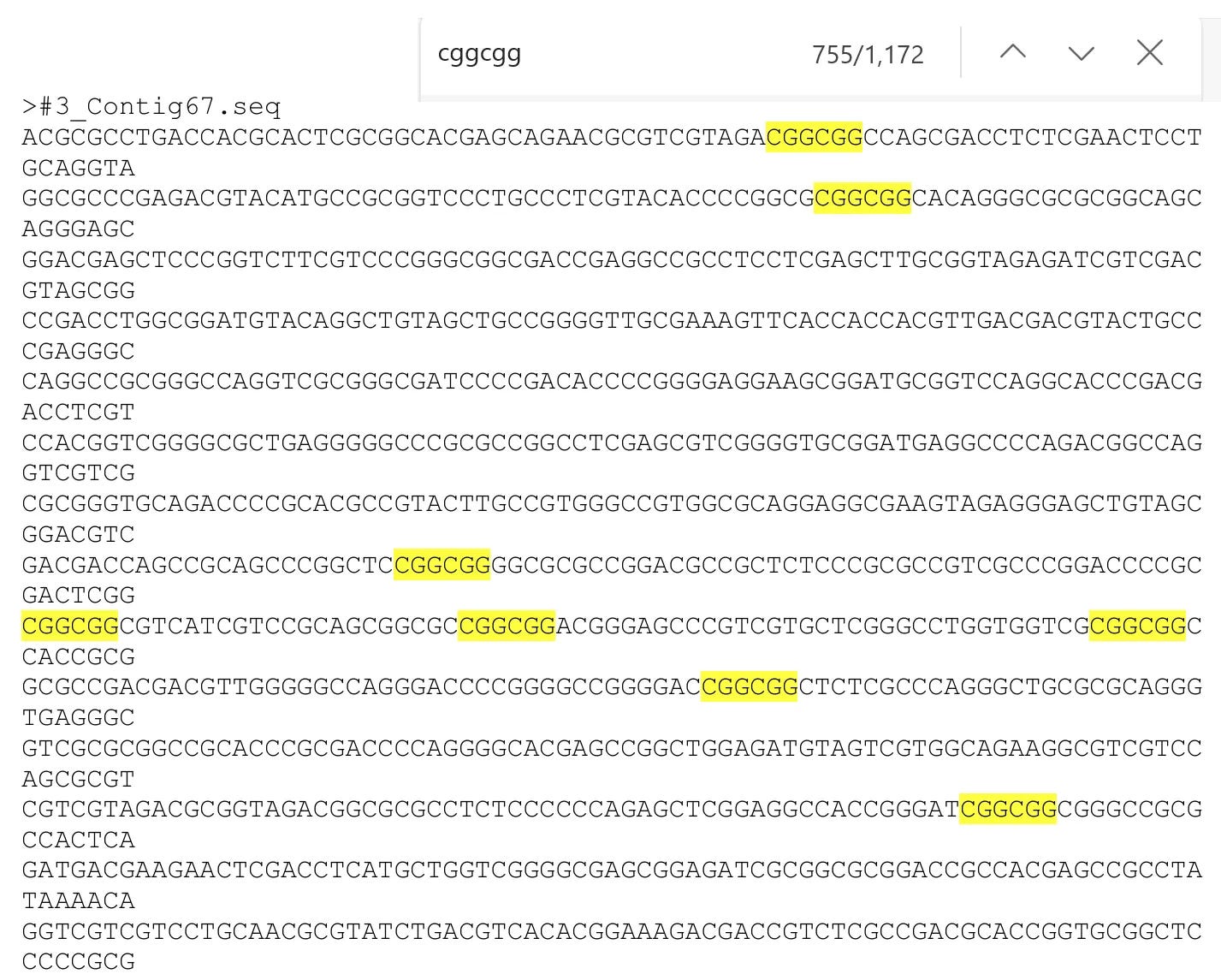

Further, they were trying to sequence another herpesvirus from a different bat species that was even more CpG enriched, but they may not have succeeded (I’ve been unable to find it in GenBank).

Herpesviruses can survive for decades in the host as a circular DNA dormant inside a cell. They can later be reactivated and replicate when another virus infects the cell. This sounds like a scenario that could plausibly result in recombination, although it is unknown for a DNA virus to recombine with an RNA virus. I don’t believe that happened, but it might be that’s what the engineers of SARS-CoV-2 want us to think, hence the peculiar codon choice.
In similar fashion I found what appears to be a peptide from a WIV discovered bat adenovirus (WIV17) substituted into the SARS-CoV-2 RBM. Adenoviruses are also DNA, and some utilize similar strategies to herpesviruses. More in a future article.
Have MERSy
There is an obvious explanation for the derivation of the amino acid sequence of the FCS, which recently became more obvious. All its reputedly problematic characteristics: the leading Proline, the suboptimal cleavage efficiency, O-glycosylation sites, the extended loop, can be easily explained by looking at the MERS FCS - which indeed most scientists interested in SARS were also looking at.
Recently A.M. Lisewski noticed in GenBank a sequence for a variant of MERS which had been passaged in mice over 30 times. This passaging resulted in only two spike mutations one of which was added an extra Arginine in the FCS.


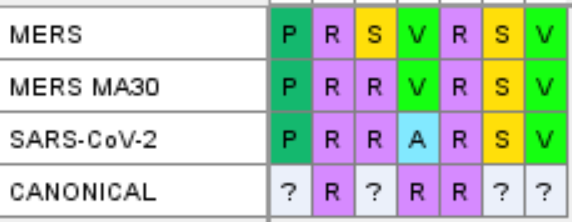
Although only a single amino acid is different, it is in the non-canonical position, the same as SARS-CoV-2. This in another extremely unlikely coincidence, an R in that position is rare. If the similarities to MERS weren’t enough already, this should have sealed it. Lisewski presented to the WHO group investigating the origin - SAGO, who weren’t persuaded.
There is only one remaining amino acid difference between MERS MA30 and SARS-CoV-2, and that V→A is a mutation a scientist would be likely to introduce in order to isolate the contribution of individual residues (a very commonly used technique Alanine scanning which replaces residues one at a time with A).
The first author on this paper was Kun Li, based in Stanley Perlman’s lab at University of Iowa - but formerly of the Wuhan Institute of Virology. His work caught the eye of Ralph Baric who sought approval to make these mutations himself and assess the effect on virulence.

I don’t subscribe to theories Baric was directly involved in engineering SARS-CoV-2, but he was being circled by people with connections to WIV and AMMS. While he wasn’t working directly with Zhengli Shi, he was a collaborator and confidante of Fang Li who, though US based, has strong ties to Wuhan. Fang Li and Zhengli Shi were in turn collaborating on a review paper with Jie Cui - who was responsible for sequencing RaTG13, and I think central to a pre-planned cover-up.

From a public health perspective, it makes little sense that there should have been such interest in SARS or MERS. The last SARS case was over 20 years ago. Despite briefly causing some localized panic, there were only ~800 deaths worldwide. A trickle of MERS cases continues, such that it has now killed more than SARS (957 at last count) but human-to-human transmission is rare. In contrast, seasonal respiratory diseases kill up to a million people every year. The scientific interest in SARS and MERS was only because they were seen as potential (or already proven) biological weapons.
If this possibility was foreseen, why is there such a taboo around asking whether SARS-CoV-2 may have been developed for that purpose?
Exercise for the statistically minded
Please feel free to weigh in with a comment especially if there are other factors needing consideration.
Calculate the probability that a four amino acid sequence…
should create an FCS motif (when inserted) and…
that motif closely resembles the one most studied by coronavirologists and…
has a rare, non-canonical Arginine residue…
is composed of rare codons and…
it somehow comes to cohabit a cell that a coronavirus infects and…
a replication error takes place such that…
it becomes inserted at just the right location in the S1|S2 junction of the spike gene to complete an FCS motif…
it survives, replicates, becomes dominant in the swarm and transmits to a new host species?
I know, right? “Nature is big - there’s a lot of dice being rolled”.
I asked Grok to help, taking into account the points above.
It’s answer: 10⁻³⁴. I don’t agree with some of its reasoning, but will try to work through it. In any case it’s small, very small.
Great analysis, many thanks for persisting.