
More evolutionary batshit
The cover-up of SARS-CoV-2's origin didn't end with Banal-52. Other recent "discoveries" seem to fill in gaps left by the earlier cover-up. But there are unresolved contradictions.

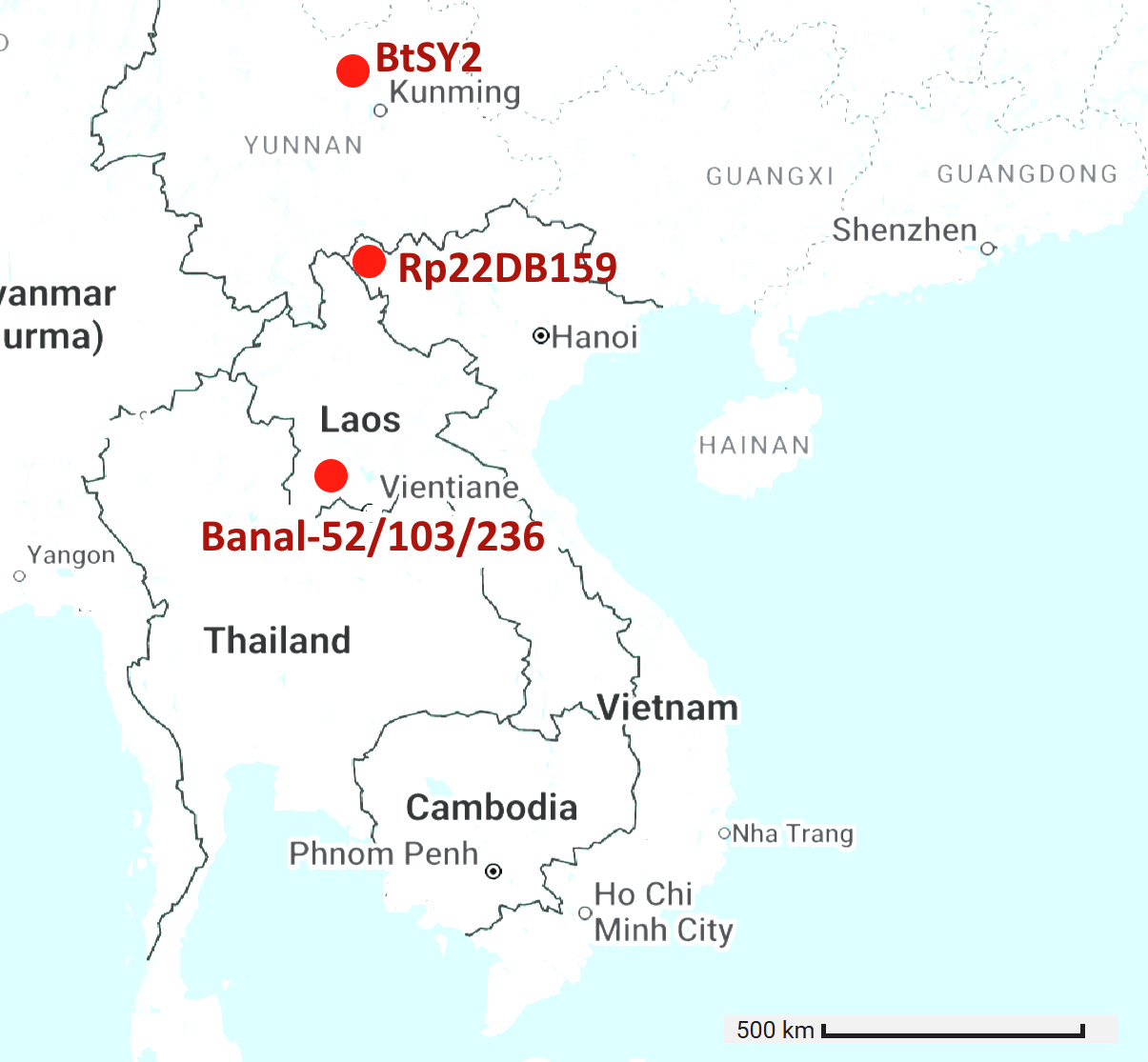
In the previous article I made the case that Institut Pasteur’s Banal-20-52 is not a natural virus but was fabricated from a chimeric sequence. But since its discovery was announced there have been more claimed discoveries of SRS-CoV-2rs, two of which have a very similar RBM to SARS-CoV-2. Today, there are 6 known zoonotic sequences with an RBM within 3 amino acids of Wuhan-Hu-1 (including 3 of the Banals).

After these 6, the next closest is RaTG13, which has 17 amino acid differences. Curiously there is no sequence with between 3 and 17 differences. Is this unusual distribution due to insufficient sampling? It seems unlikely - thousands of bats have now been sampled.
On September 20th, 2021, China’s National Health Commission lab submitted a preprint to ResearchSquare. This was a comprehensive survey, for which they had taken 17,000 samples from more than 13,000 bats at 703 locations across China between 2010 and 2021. Stunningly, they reported finding no trace of SARS-CoV-2 related viruses. They even returned to the abandoned mine in which WIV claimed to have found RaTG13, and declared it was no longer present. It appears part of a coordinated attempt to confine the origin of Covid-19 (whether natural or artificial) to be outside China - and was likely timed to coincide with the announcement of the Banals, coming just one day after they were published.
This pre-print upset some Western scientists who had previously collaborated with Chinese state and military groups on the pangolin coronaviruses and RmYN02. These results seemed to contradict their own.

But it seems the Propaganda Department relented, perhaps judging it wiser to keep their Western collaborators/promoters and prestigious science journals on-side. Soon after the controversy blew up in Science, a group from Sun Yat-Sen University reported sequencing another SARS-CoV-2 like virus, BtSY2, from Yunnan bats. Eddie Holmes got his name on another Nature paper.
BtSY2 has serious deficiencies with its data. The genome can’t be assembled from the sequencing reads. There is no read coverage at all of the RBM.
Meanwhile, the NHC’s comprehensive survey was resubmitted to a low-profile journal in which they defended their work somewhat meekly. The contradiction was never resolved.

Hughes and Holmes seem well aware that this paper likely has a propaganda purpose. They also understand that Chinese scientists are compelled to follow an official narrative. But they are unable to recognize that their own collaborations with Chinese state institutions might similarly be tainted by fabricated data. They haven’t been involved in generating that data (i.e. sequencing), merely helping to interpret it. While Hughes collected samples in Yunnan, she sent them to Shandong for processing - to a scientist who works closely with AMMS. There is no way for her to know what her samples really contained. But their names lend the papers credibility and help get them published in prestigious western journals.

The success rate for sampling SARS-CoV-2rs varies dramatically. WIV claimed to have found RaTG13 in one bat in a Mojiang mine shaft, but several other surveys reported nothing of interest at the same site. Institut Pasteur found 7 SARS-CoV-2rs (2 are incomplete and unpublished) in one cave in Laos, but none at another 3 sites. In Vietnam, Laos and Cambodia, Institut Pasteur has reported discoveries at a number of sites - some very close to the Chinese border. Why were none found in the first 15 years of hunting for bat sarbecovs, but since 2020 they have been reported across a vast expanse of China and South-East Asia?
My suggestion is there may be no such thing as a natural SARS-CoV-2 like virus. Every set of sequences presents evolutionary anomalies and/or has issues with sequencing data. Most have links to the same closely related parties - WIV, AMMS, Institut Pasteur. We should not unquestionably trust any sequence related to these events.
More batshit examples…
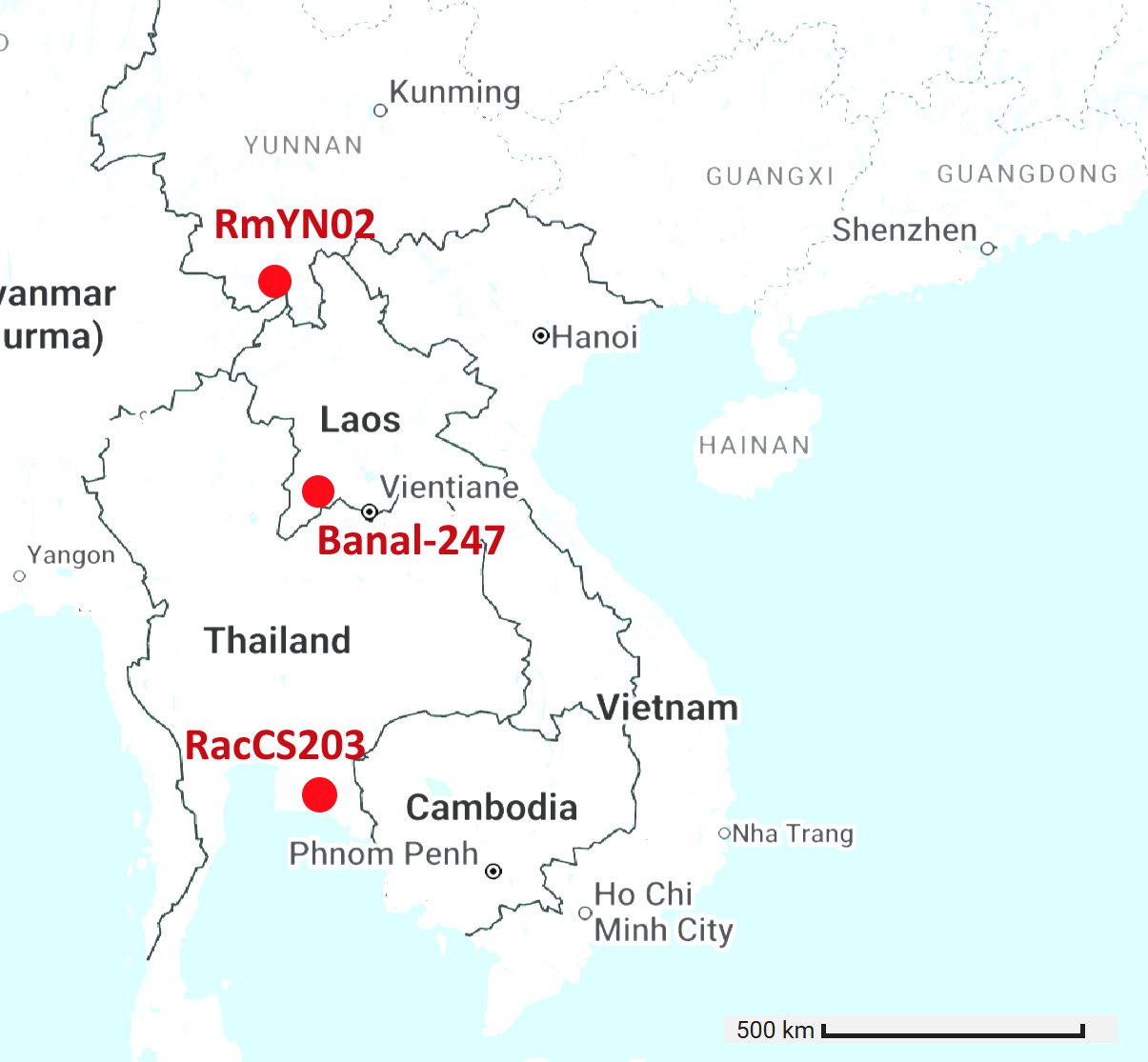
The locations of the 6 viruses with RBM closest to SARS-CoV-2 are plotted below (GD pangolin cov is excluded as its origin is unknown). There is ~900km between Banal-52 and BtSY2. That’s a long distance for tiny horseshoe bats to fly, they typically migrate distances of 20-30 kilometers. Given the RBM is expected to be the most variable region of the genome, there are astonishingly few differences between the bat viruses, and also human and pangolin viruses.
Banal Sequences
The 5 Banal viruses can be divided into three groups:
SARS-CoV-2 like: Banal-52 (which was described in an earlier article).
GD Pangolin CoV-like: Banal-103 and Banal 236. These are 98.4% identical to each other through the genome.
RmYN02-like: Banal-116 and Banal-247 which are 99.9% similar to each other, but hint at some interesting evolution.
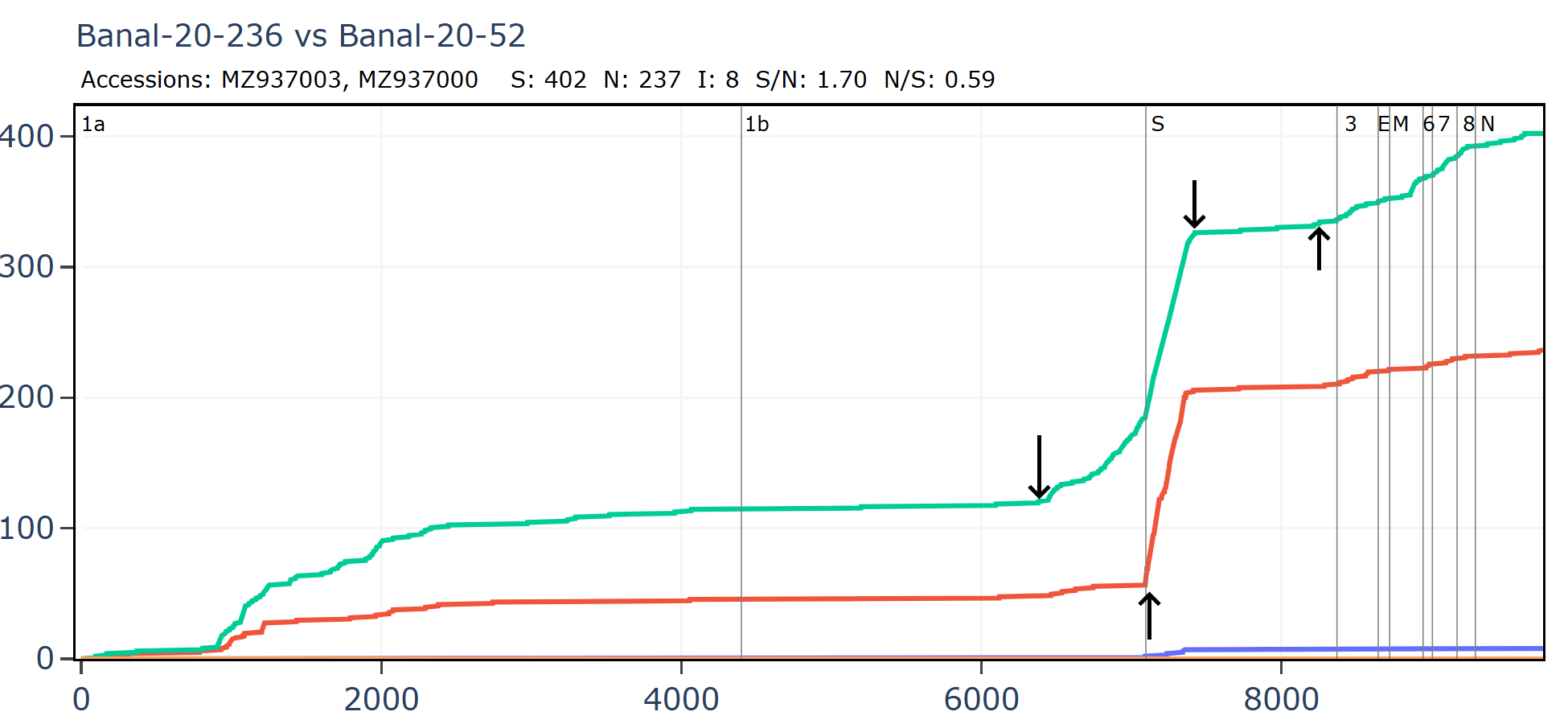
Banal-52 and Banal-236 are very similar for much of the genome, aside from an apparent recombination in which the NTD of spike is replaced.
The bat/pangolin connection
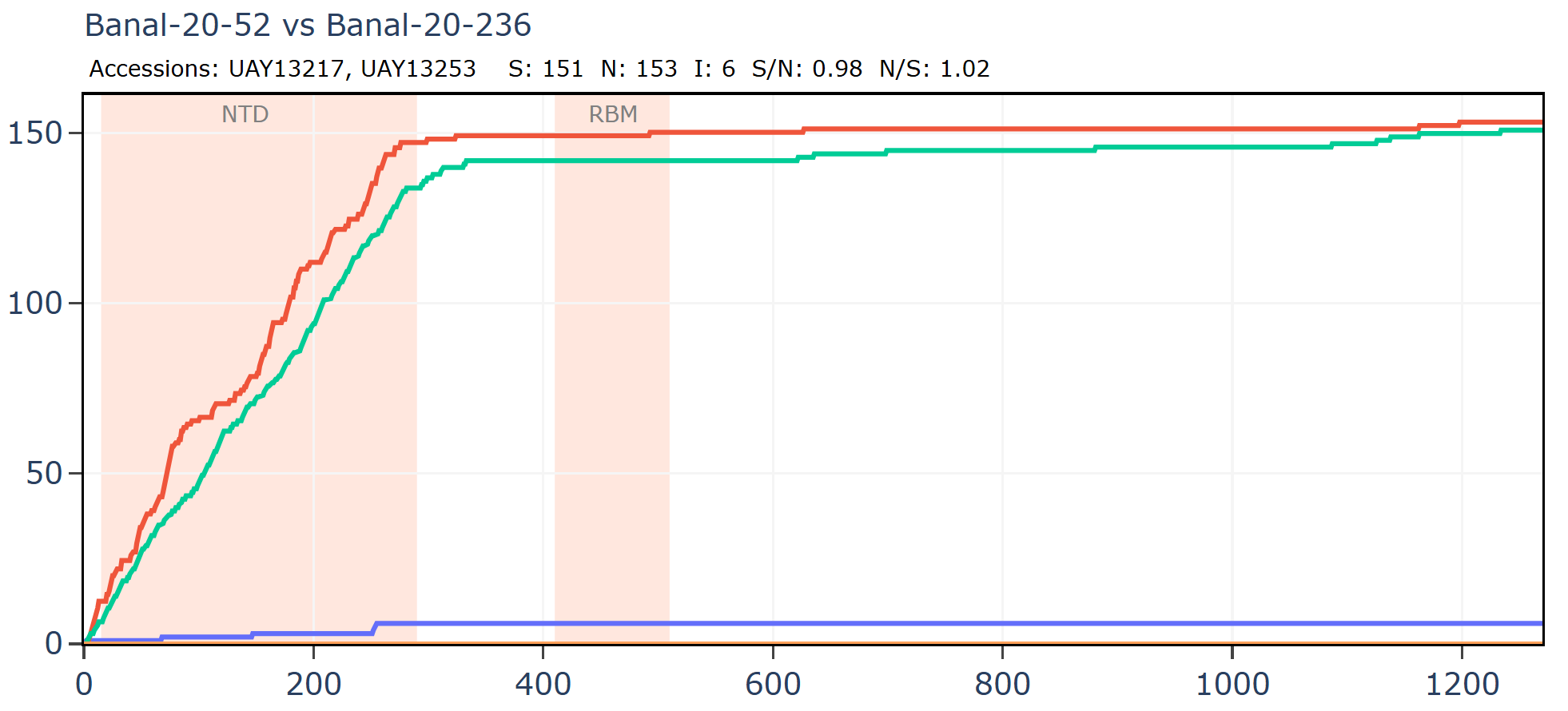
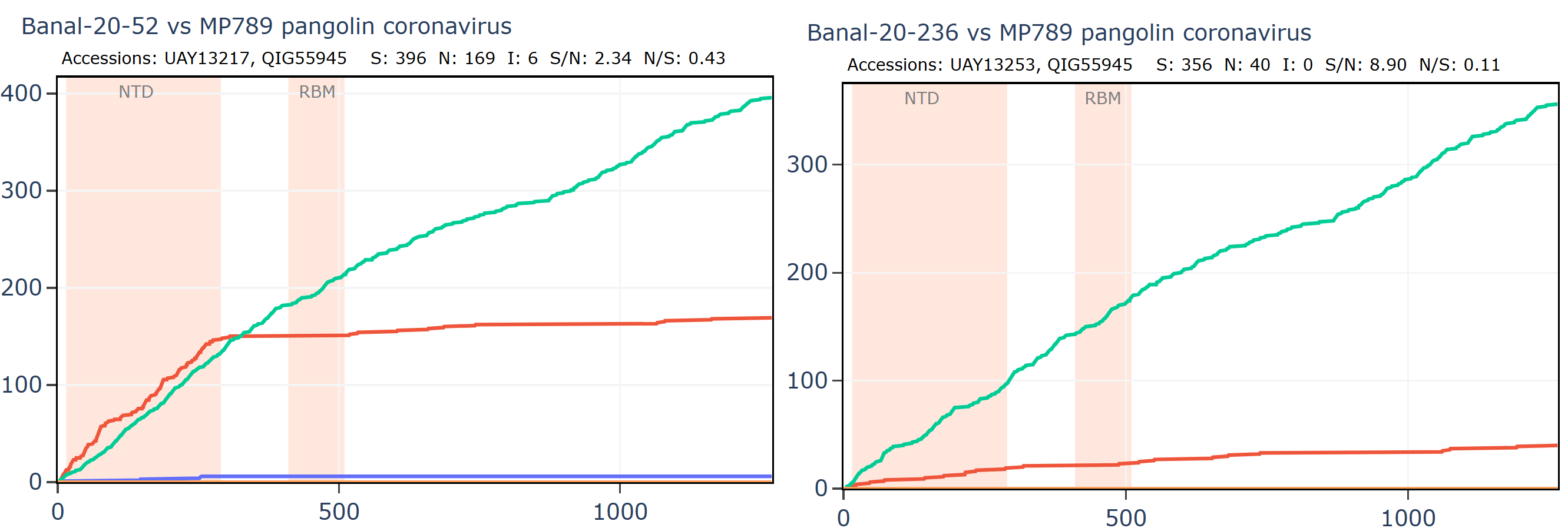
Banal-236 is closer to the GD pangolin coronavirus in the NTD. The ratio of synonymous to non-synonymous mutations at ~9 is extremely high between Banal-20-236 and MP789 particularly considering a host and system tropism change.
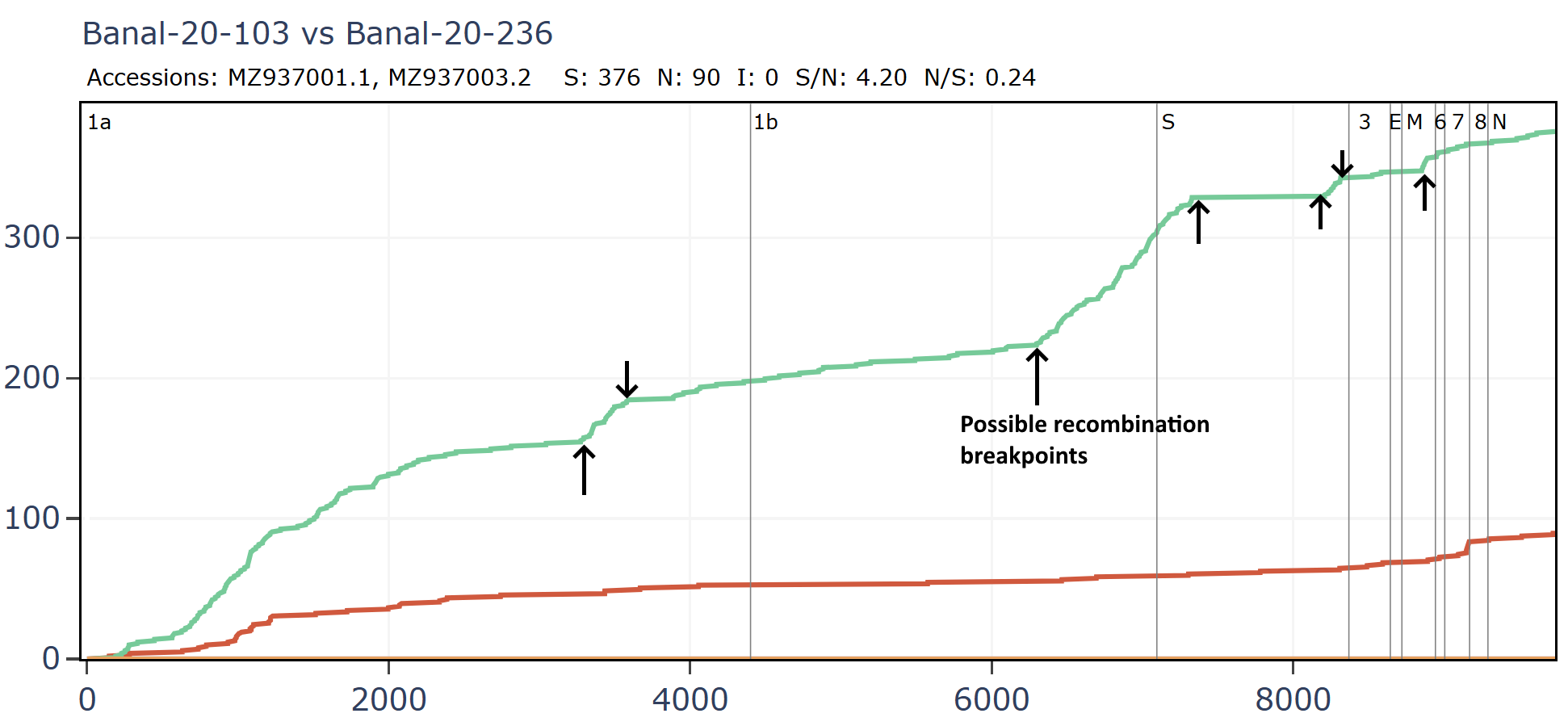
Banal-20-103 and Banal-20-236 are very similar in the spike, partly because of another apparent recombination. This can be inferred visually by a clear change in the rate of mutations. A sharp change in gradient of the synonymous (green) line is most telling. The slope of the non-synonymous (red) line often changes because of differences in selection pressure in different regions, and may not always point to recombination.
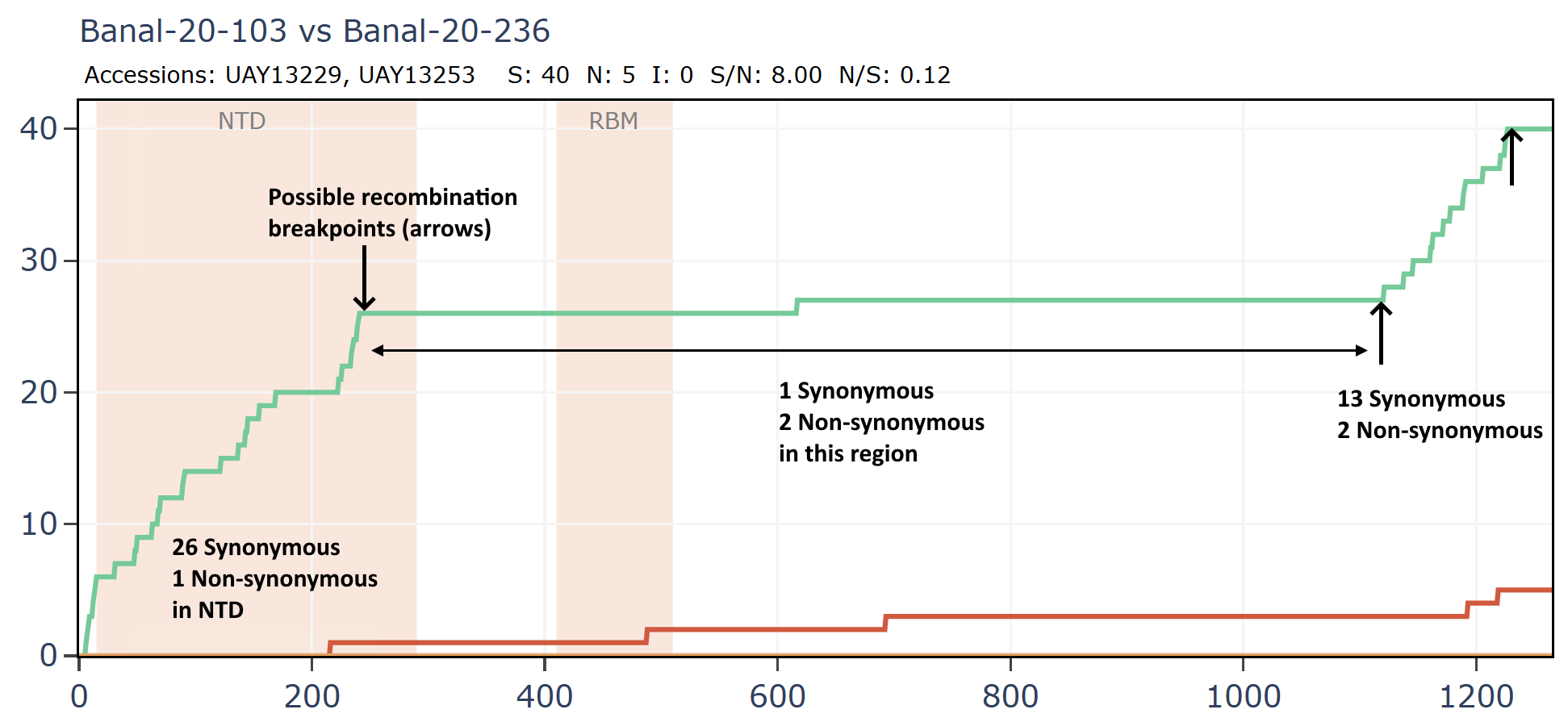
There are several possible recombination points in the full-length genomes.
RmYN02 also validated?
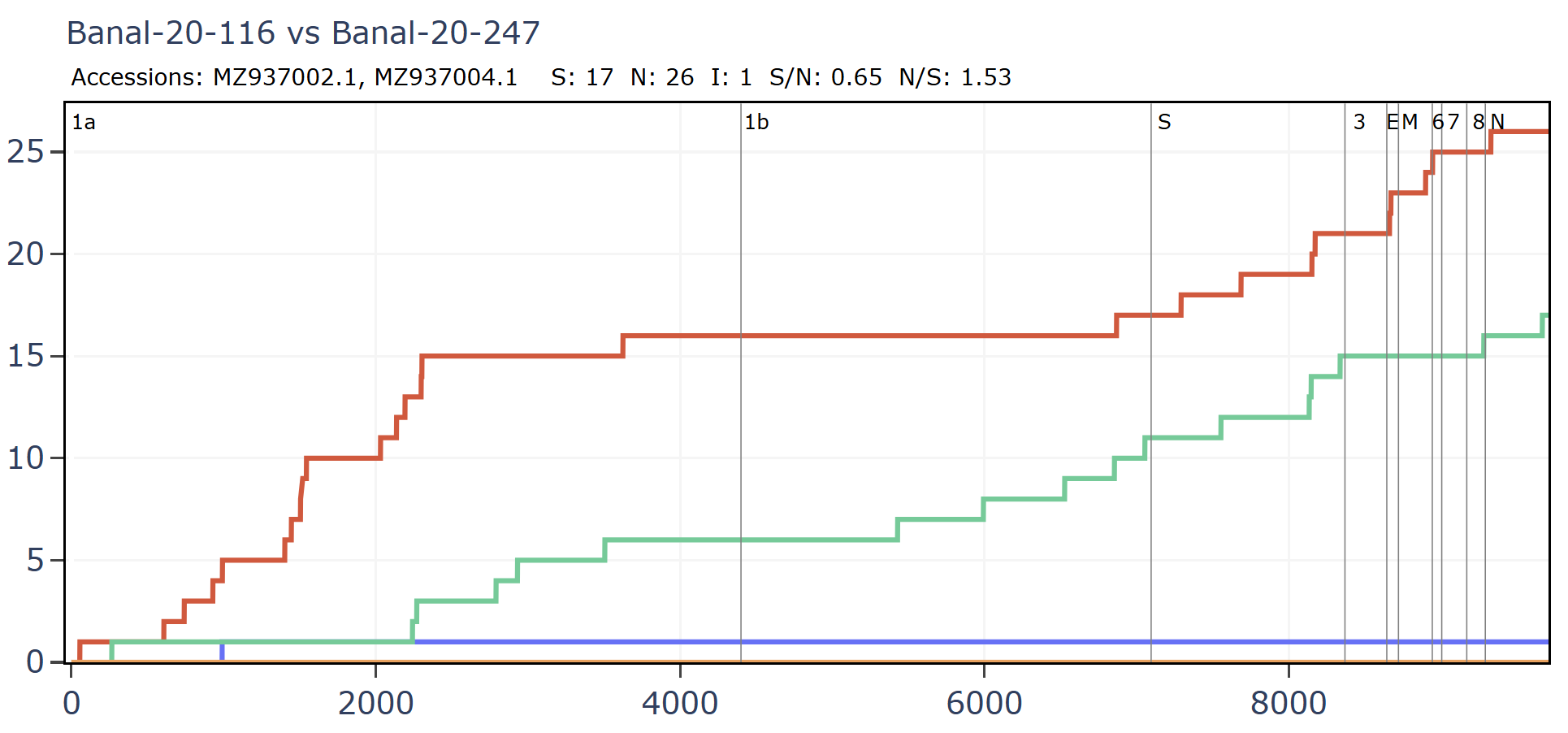
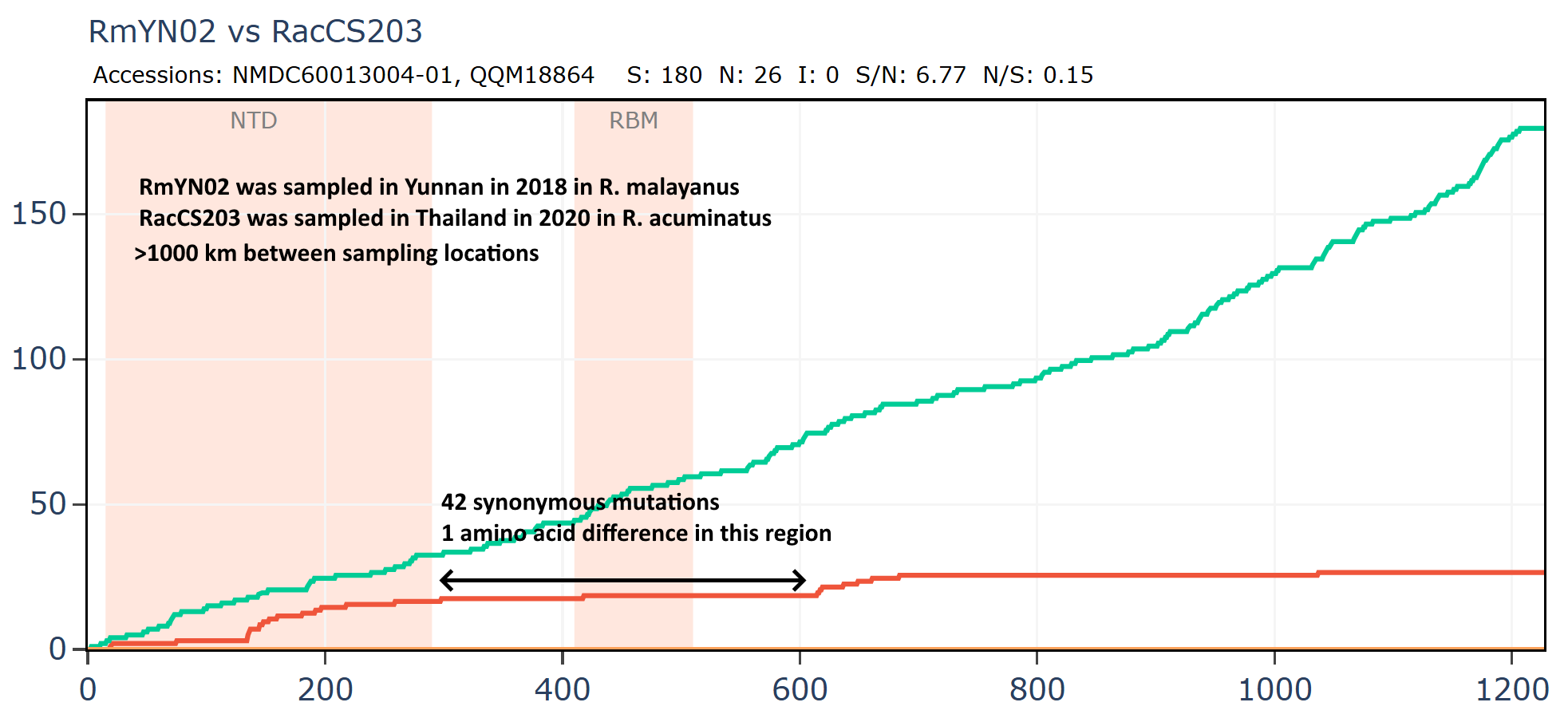
Banal-116 and Banal-247 are a different species to the other Banals, though extremely closely related to each other with ~99.9% nucleotide identity. They are also closely related to RmYN02 and a coronavirus from Thailand, RacCS203. Although most authors are from Thailand, Tu Changchun from AMMS and Linfa Wang were also involved in sequencing these viruses.
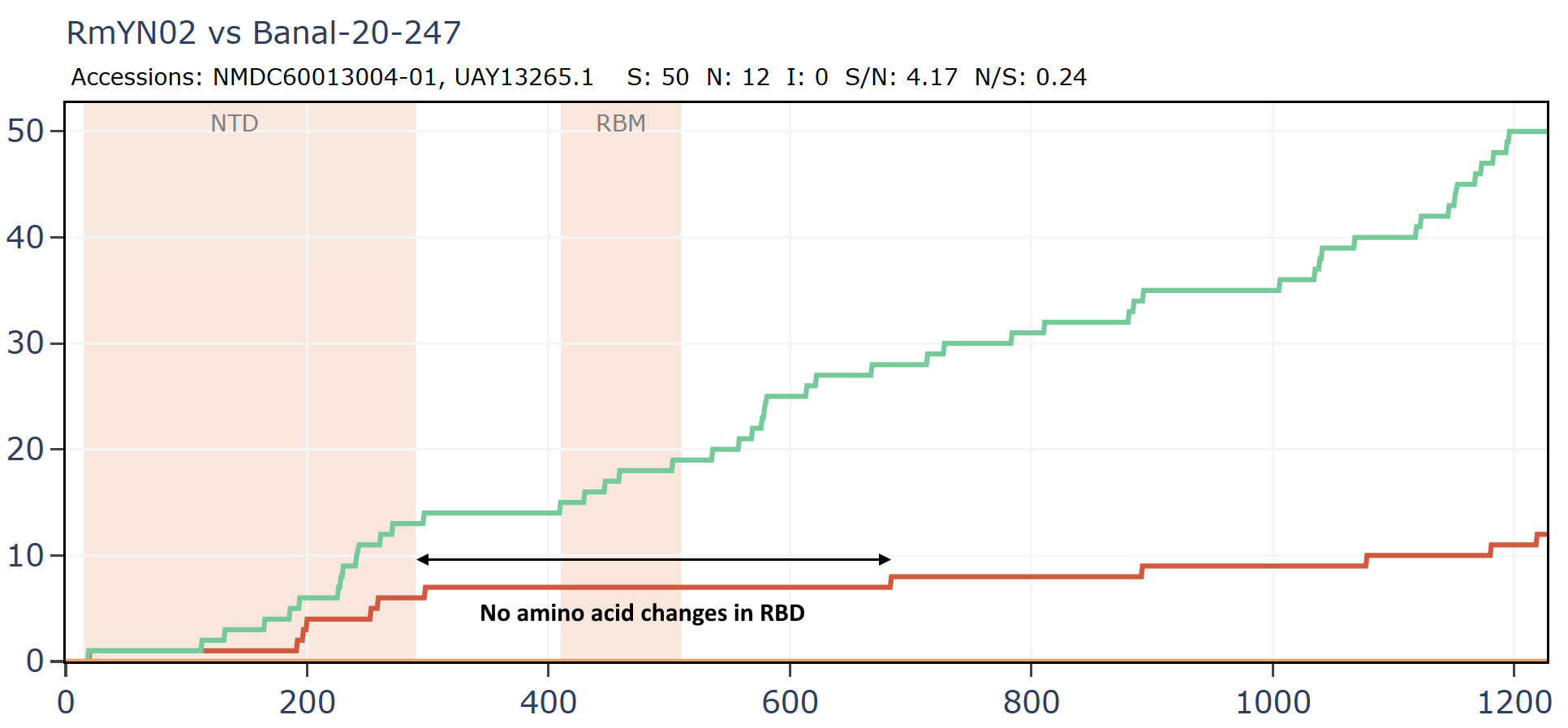
RmYN02 and Banal-20-247 have no amino acid differences in the RBM, or extended RBD although there are 13 synonymous mutations in that region.
Banal-247 and RacCS203 have just 1 amino acid change in an extended region around the RBD, while there are 55 synonymous mutations.
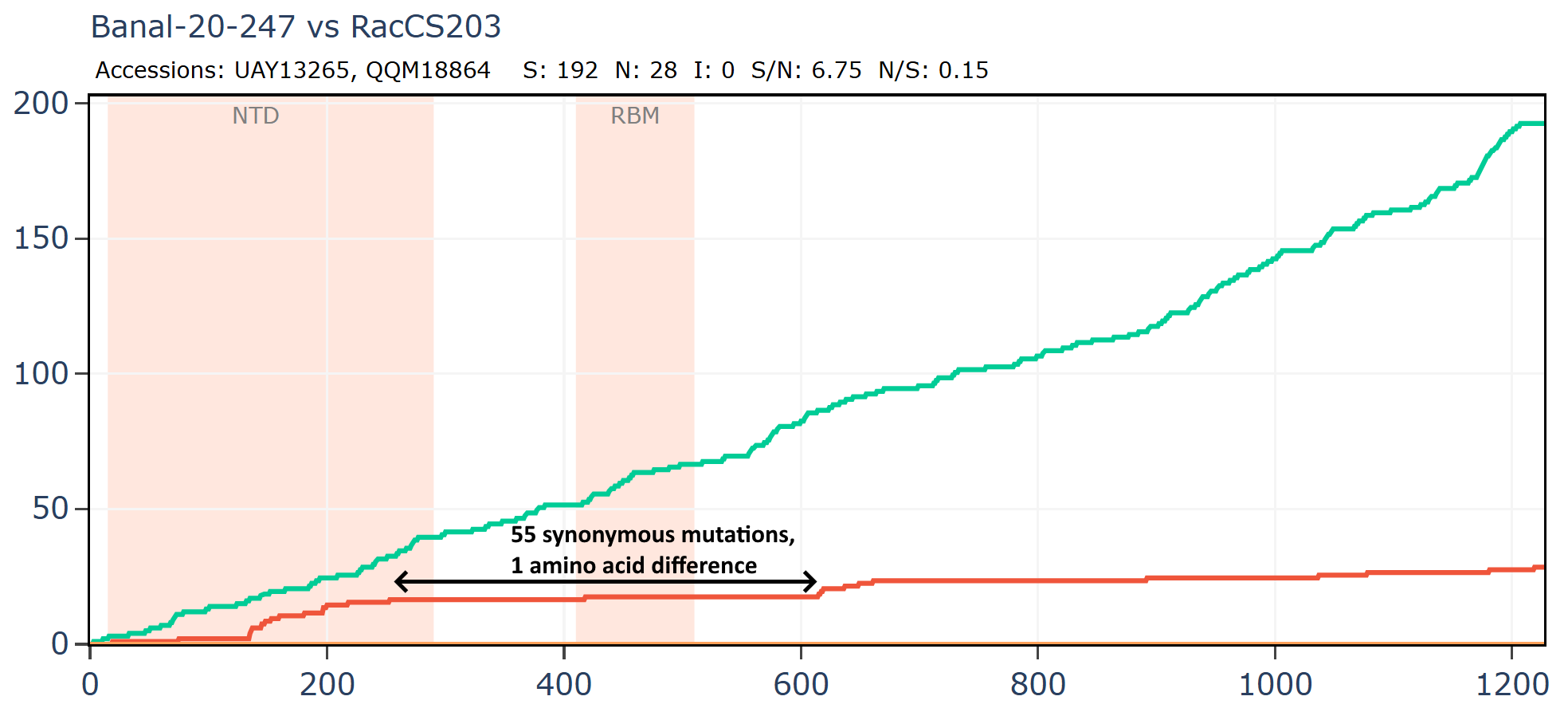
Banal-247 was sampled approximately 500km distant from RacCS203, RmYN02 is 1000km distant from RacCS203. Interestingly, while sampled much further apart, the latter pair are closer in spike sequence with 206 total mutations vs 220.
Filling in the gaps - Hassanin et al.
Interestingly BtSY2 is claimed to have Tyrosine at 501 which is a most significant RBM mutation in human SARS-CoV-2 variants, having emerged independently several times. BtSY2 also has doubts over its provenance, it isn’t possible to assemble the genome from the raw read data provided. More about this in a future article.
Nonetheless BtSY2 has a curious recombinant relationship with Rp22DB159, another of the set from Vietnam sequenced by Institut Pasteur. These were collected four years apart (2018 and 2022), but also at a considerable distance (~400km). The N501Y mutation is the only amino acid difference in a region over 850 amino acids.
Frozen evolution
Cambodian virus RshSTT200 from Institut Pasteur Cambodia has a very odd relationship with a virus discovered recently in Vietnam, Ra22QT137 part of a set which were also sequenced (mostly) by Institut Pasteur. These two samples were collected ~450km and 12 years apart (in 2010 and 2022). In the first 450 amino acids of spike there are just 5 silent mutations and no amino acid changes - an extremely slow rate of mutation given there is a bare minimum of 12 years since they split from a common ancestor. In the middle of the RBM the rate of mutation increases substantially, suggesting a recent recombination with a species with a divergent S2. But how - since one of them was stored in a freezer?
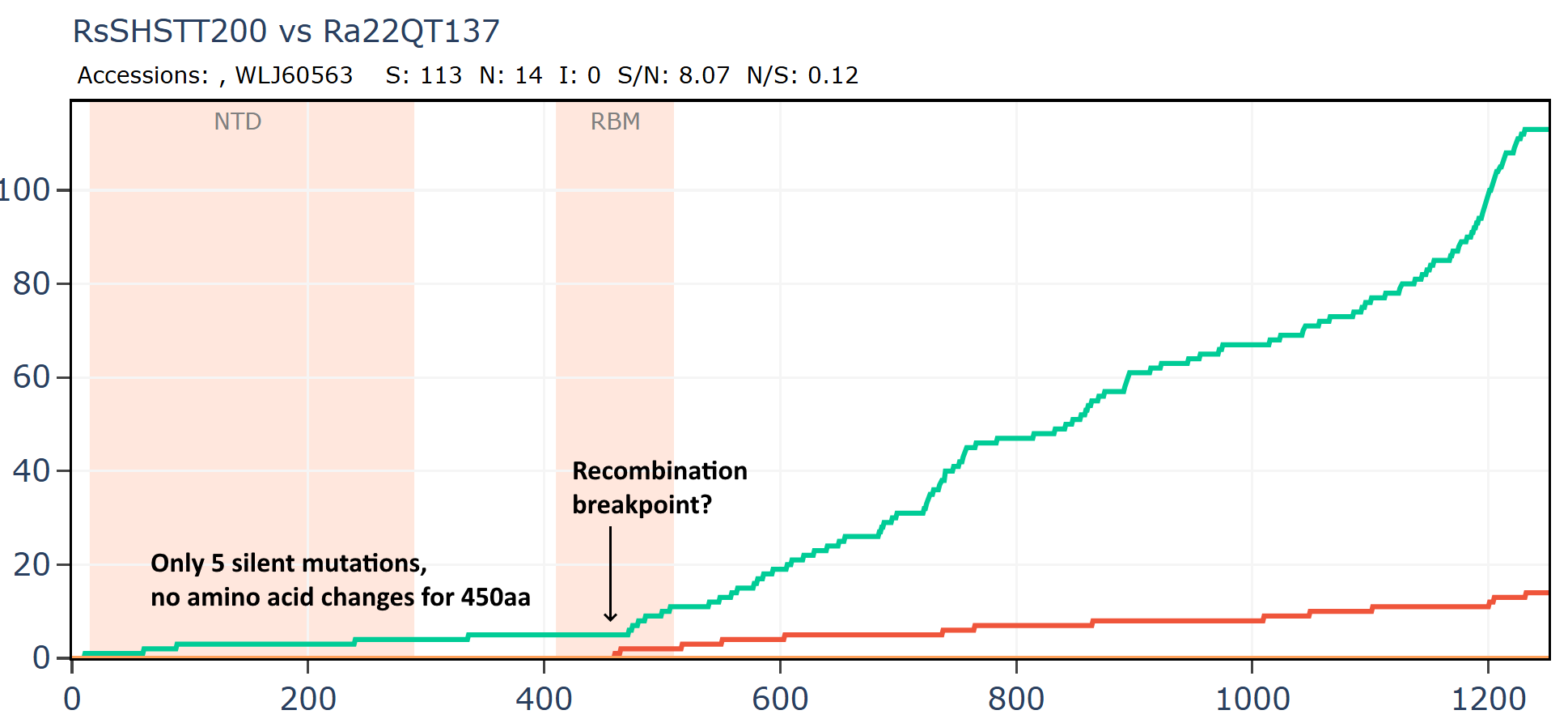
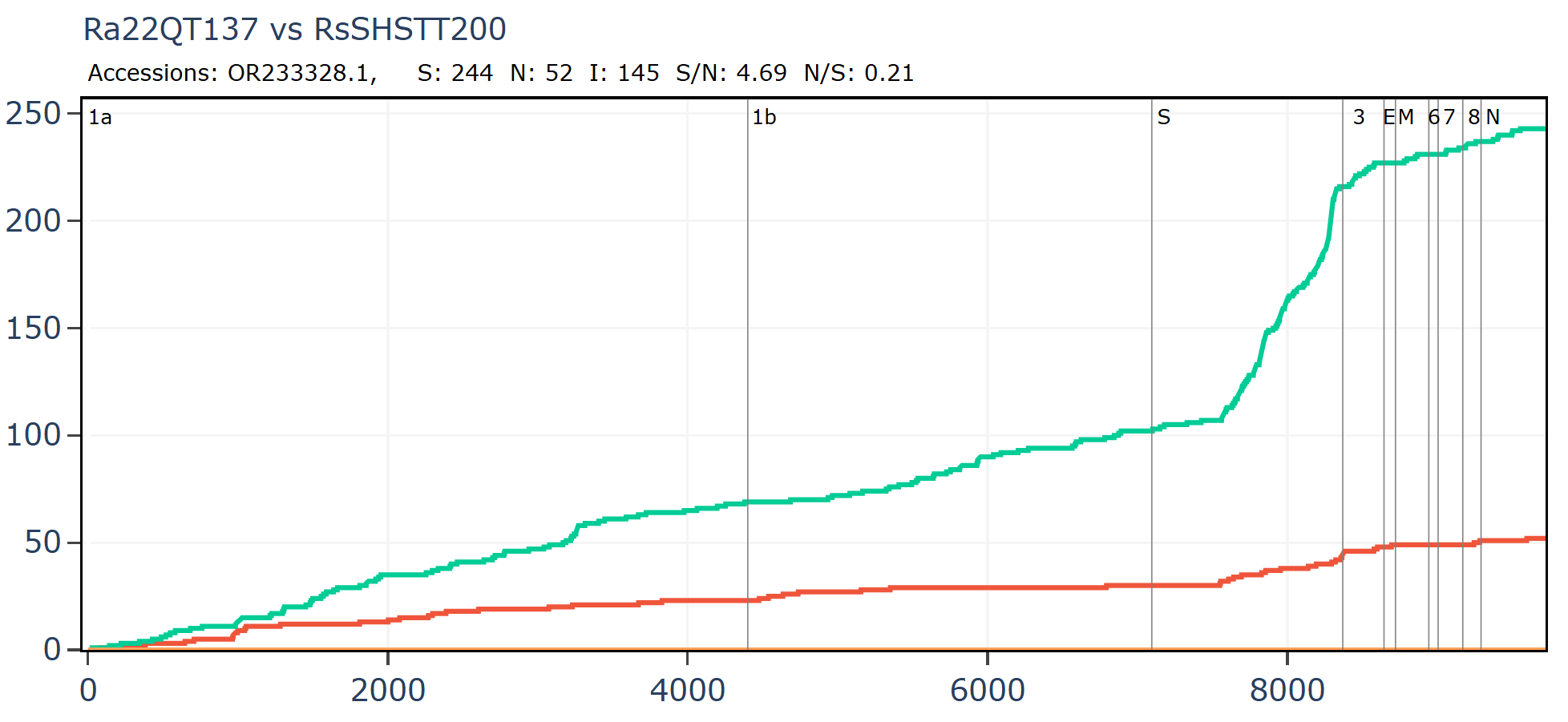
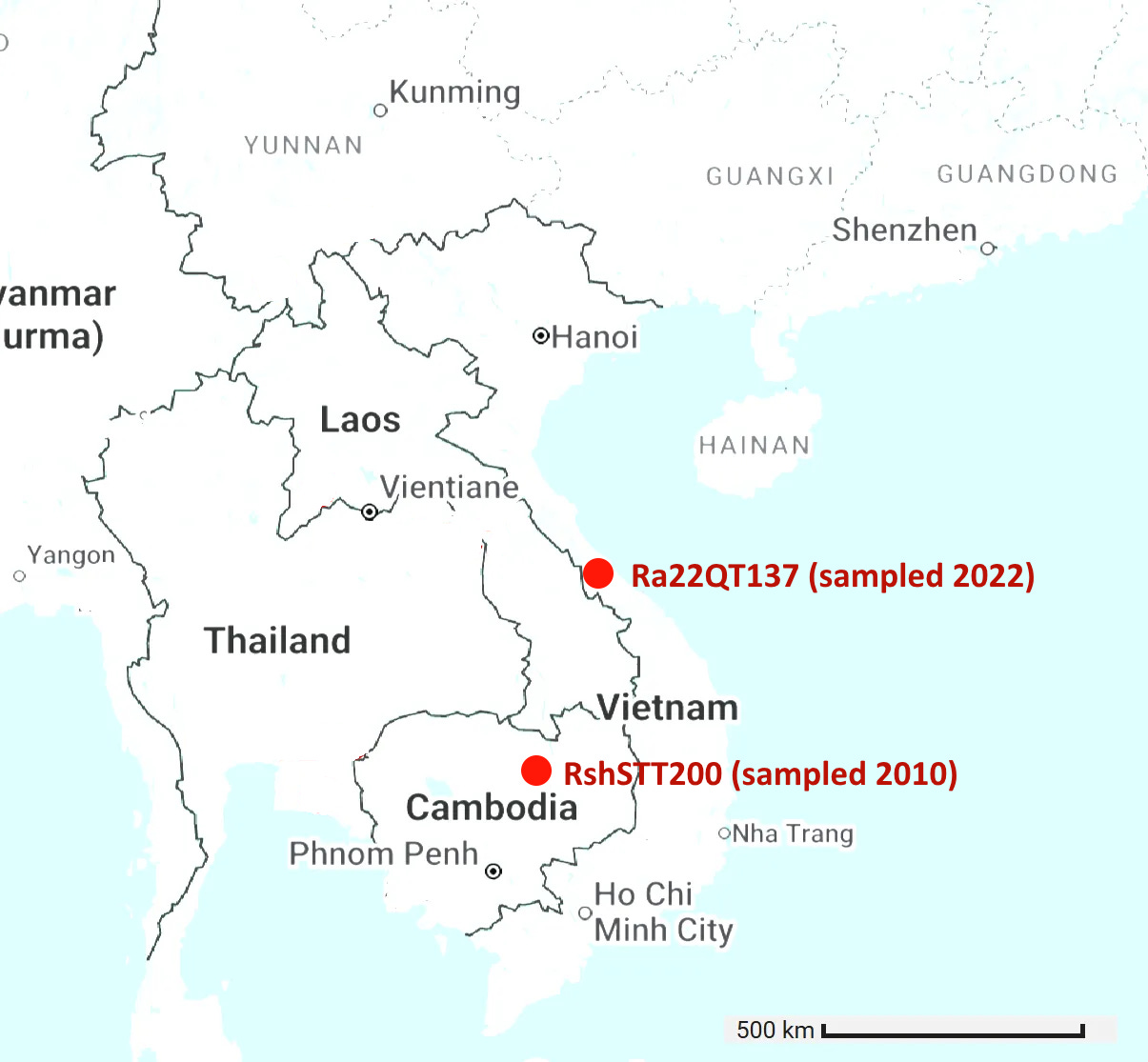
An RBD frozen for 18 years
Another virus from this Vietnamese set Rt22CB395 is unusually close in amino acid sequence to Rp3 (the first SARS related virus discovered by WIV in 2004). The samples were collected 18 years apart - in 2022 and 2004 respectively, yet the RBD is unchanged. The ratio of S/N mutations is also very high.
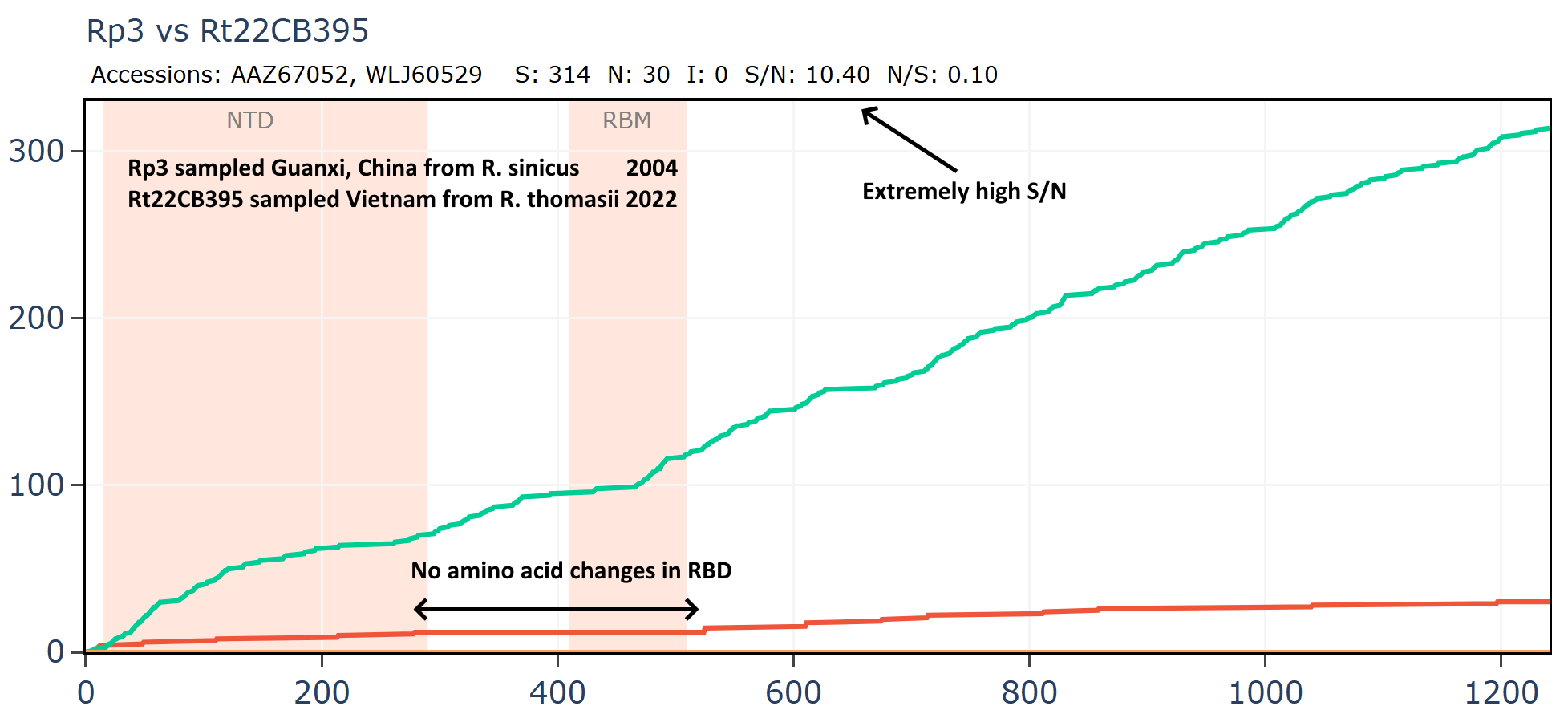
But if we compare Rp3 to another virus collected in 2004, HKU3-1 it is not only overall more distant but has much lower S/N ratio and has 14 mutations in the RBM.
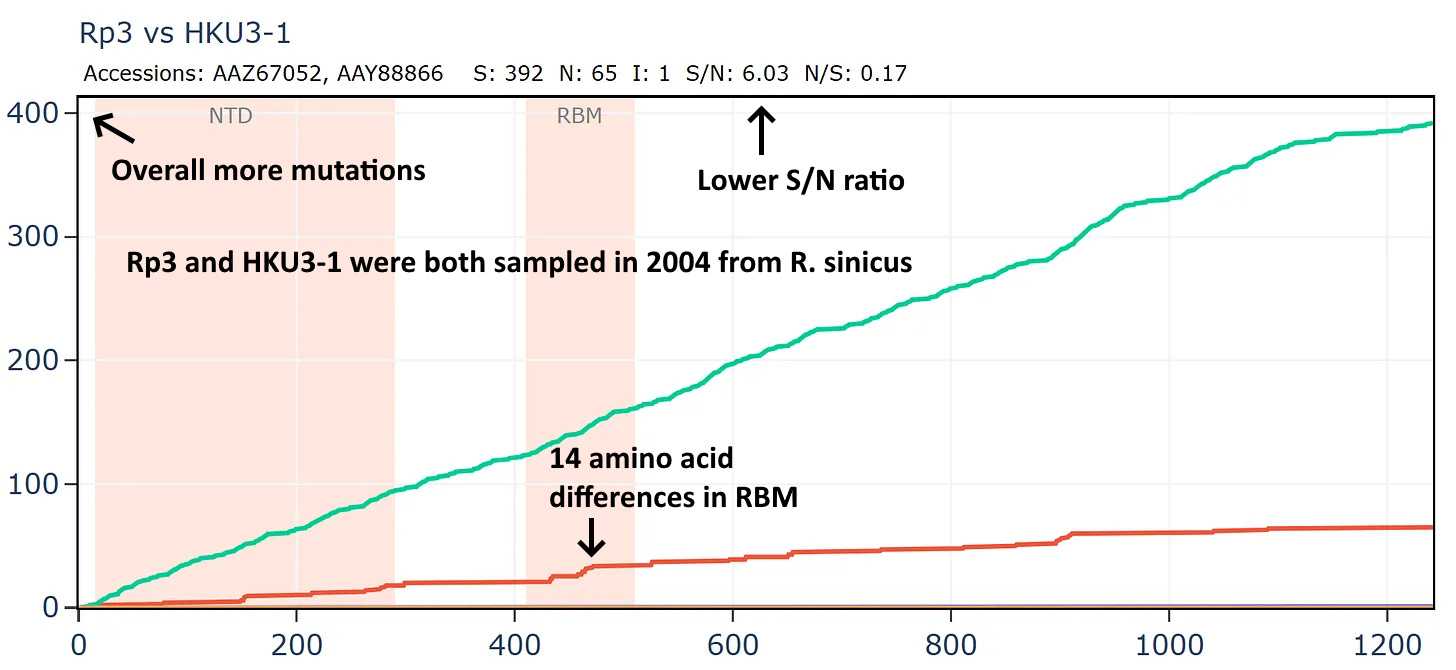
And comparing to another virus Rm1, also collected in 2004.
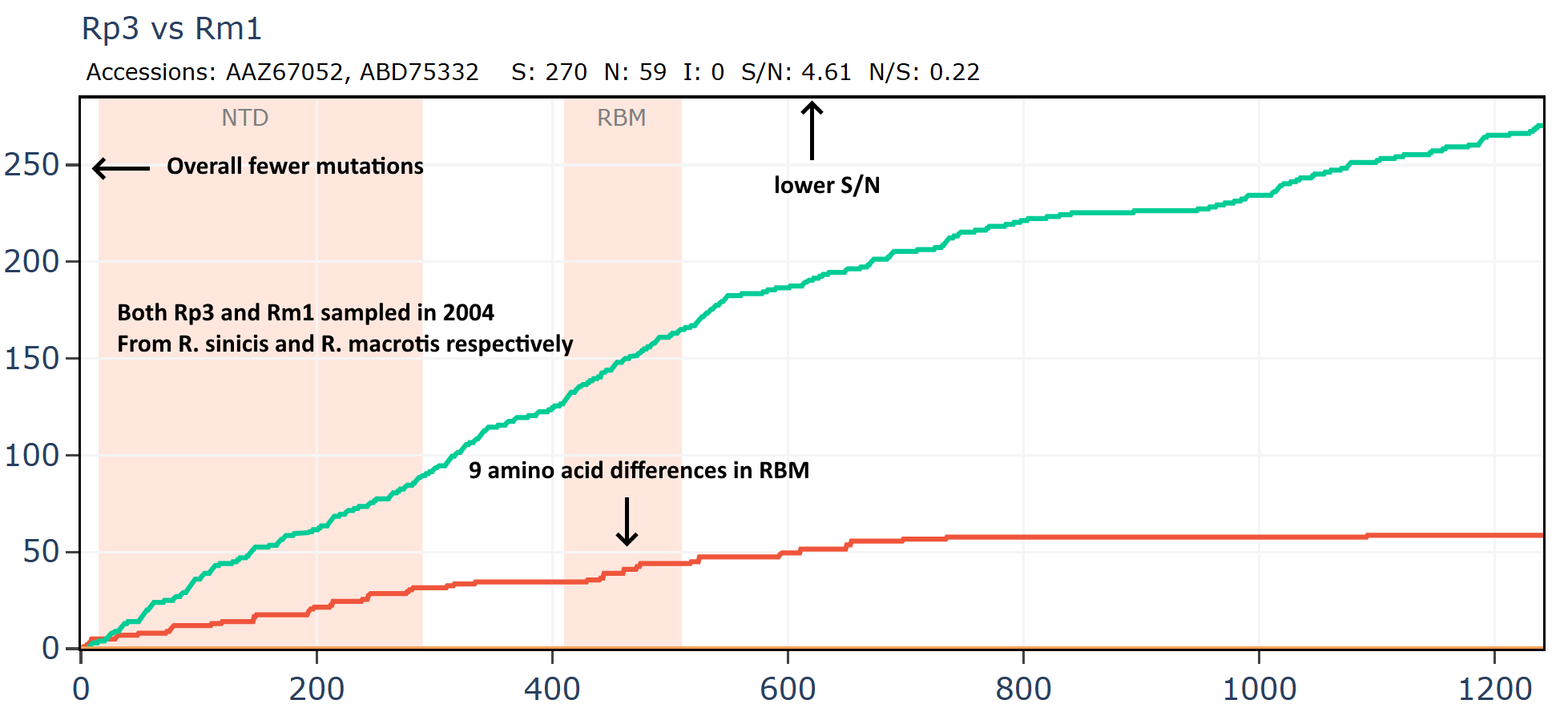
And another:
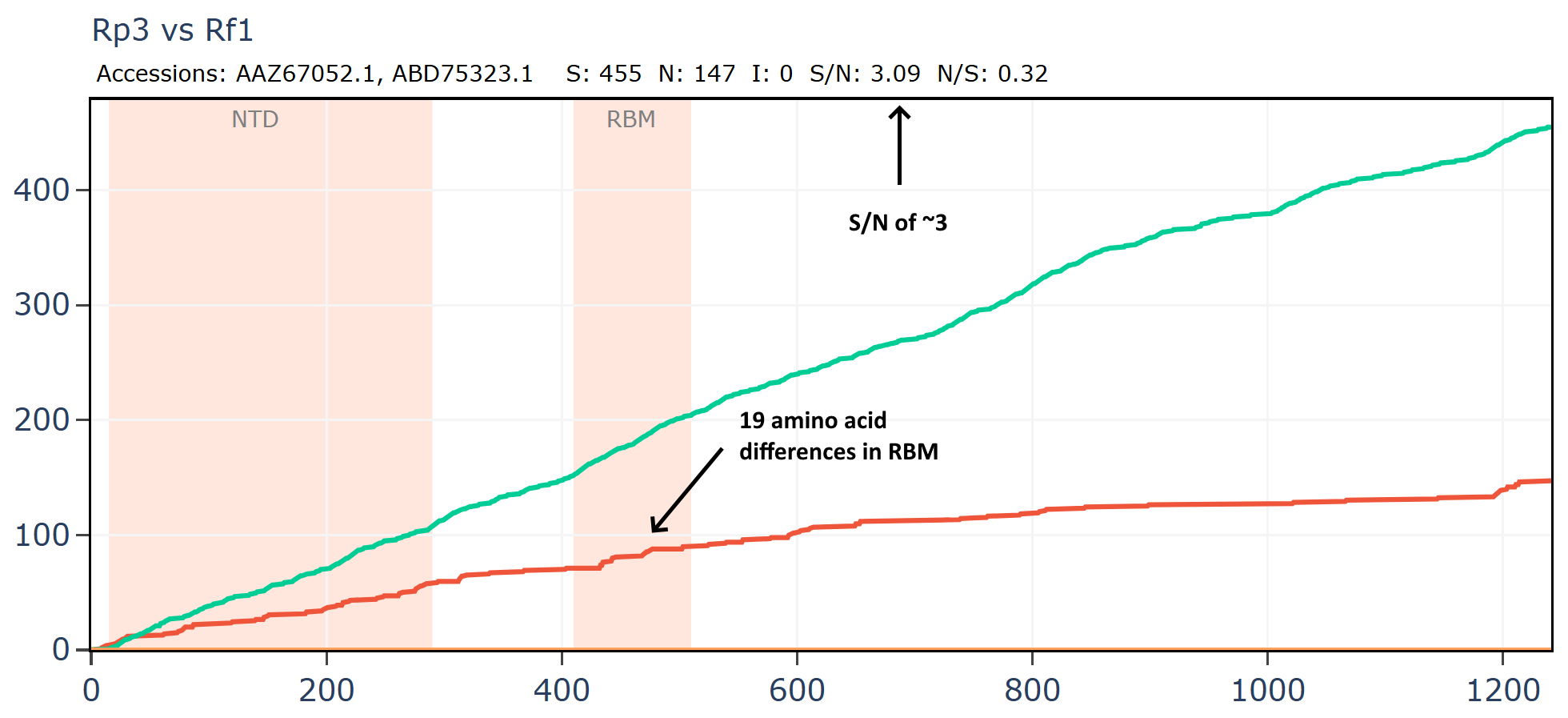
How is it that viruses collected 18 years apart in different species are more similar than those collected at the same time? Why is there no evolutionary pressure on the RBM of Rt22CB395 such that it is identical to Rp3 after 18 more years evolving?
Hyper-recombination
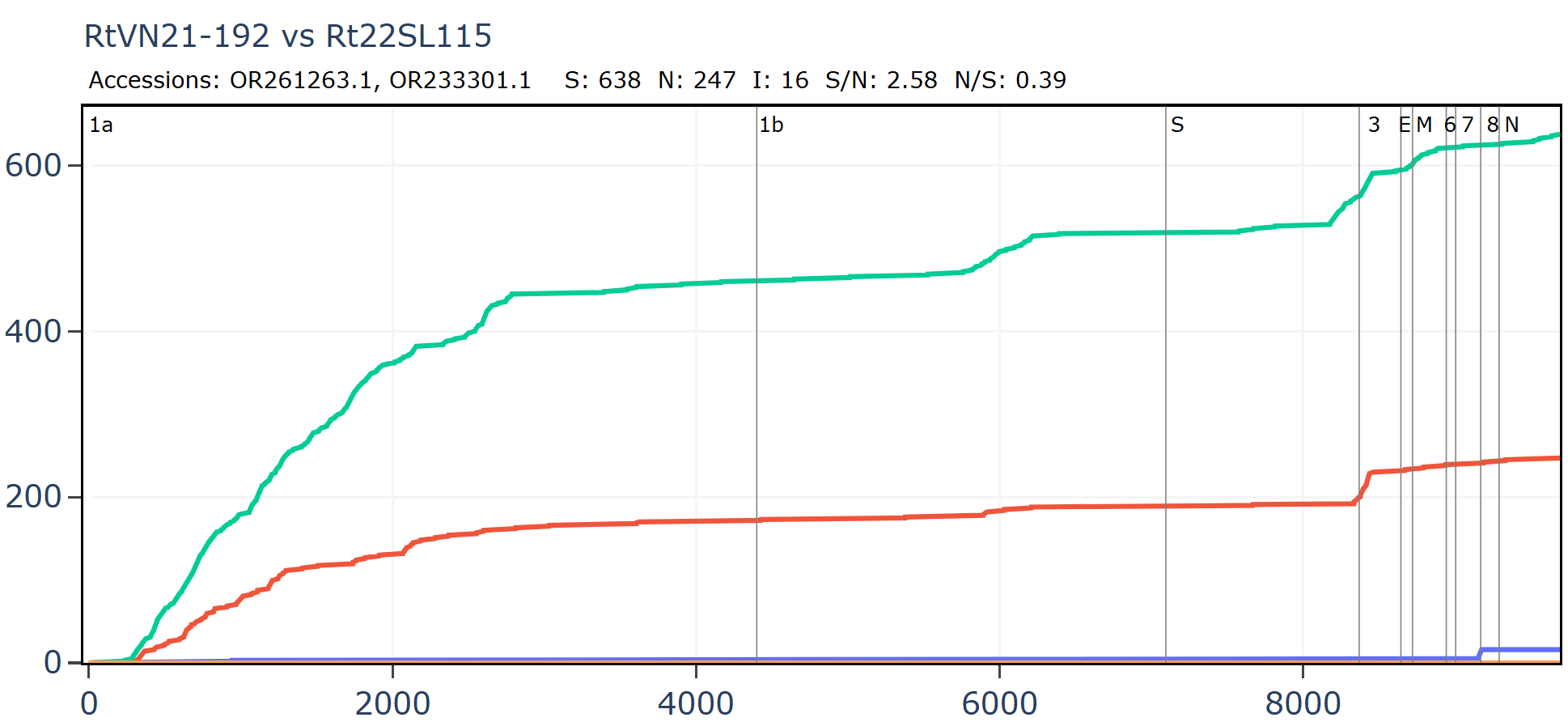
Another bizarre pair from two separate Institut Pasteur related groups, both sampled in Vietnam. In this case the samples were collected only 20km and 1 year apart in the same species R. thomasii. If they were close in sequence, it shouldn’t be too surprising. But the detail still appears quite unusual. There is just 1 synonymous mutation in the first 500 amino acids. Then the rate picks up a little, flattens out again, before a flurry of only synonymous mutations - 30 in total - at the 3’ end.
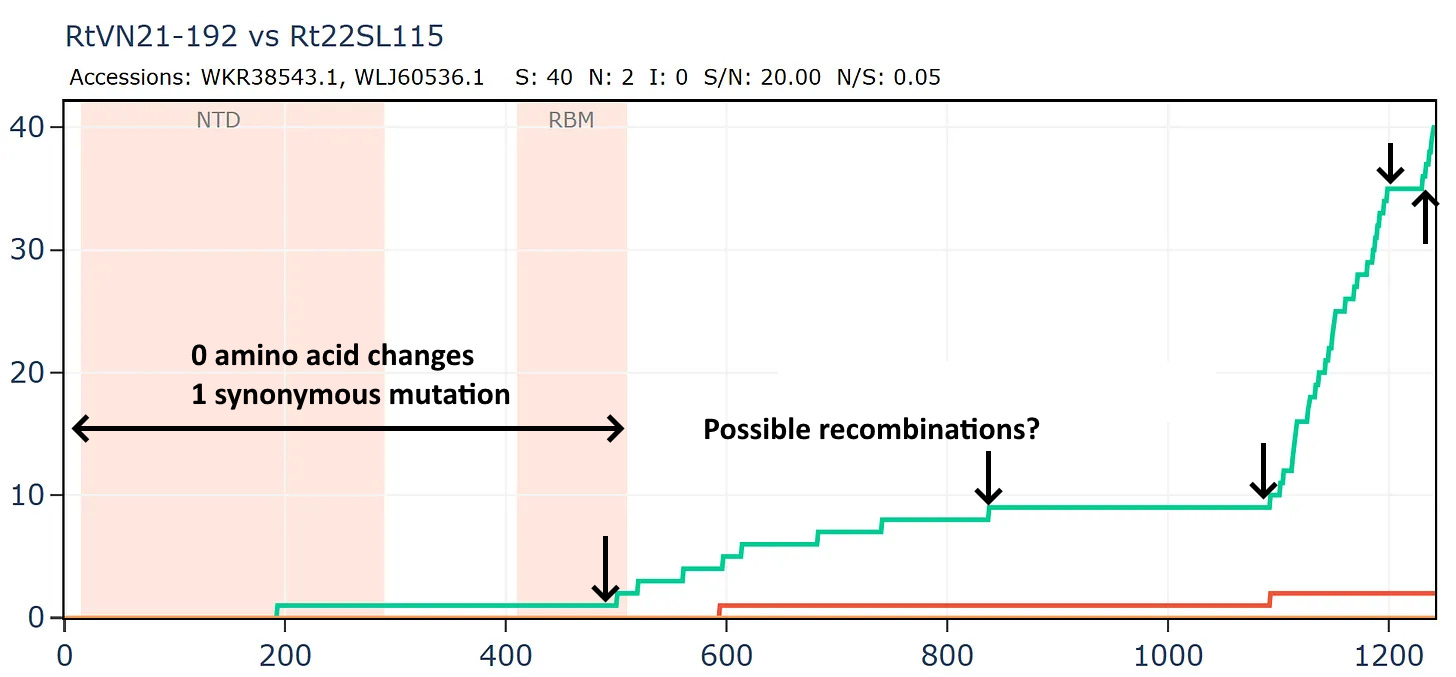
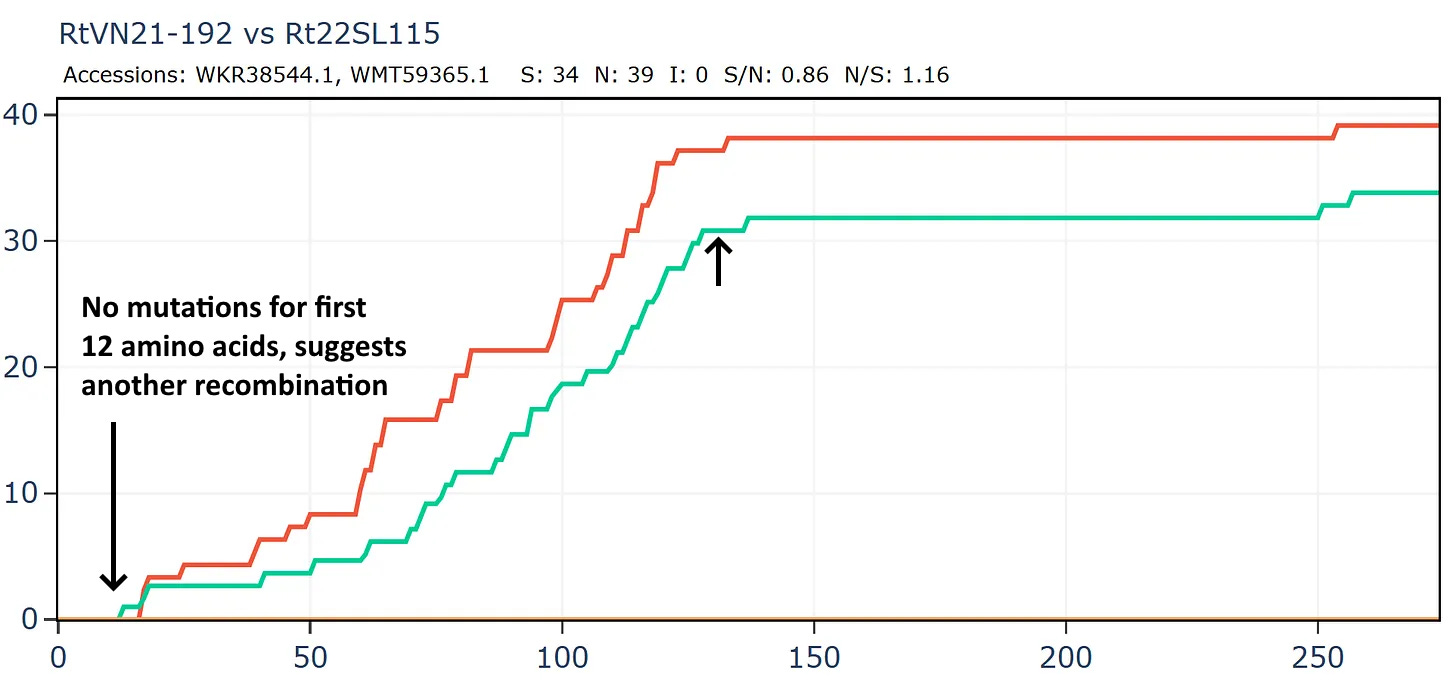
The full-length genome shows the main region of amino acid divergence is in ORF3a, which is very unusual.
But this isn’t a simple continuation of the previous recombined region, because the divergence only start after amino acid 12.
Discussion
Many sequences published since 2020, which purport to explain the evolution of SARS-CoV-2 do not conform to a conventional model of viral evolution in bats.
Common discrepant characteristics of these viruses are:
An extremely high rate of recombination. Occasionally there appear to be even more recombination breakpoints than regular amino acid substitutions.
An extremely low rate of mutation in the RBM. The RBM is expected to be a region of hypervariability as:
immune-exposed, surface location at tip of spike forces mutation to avoid antibody recognition
protein structure accommodates mutations - flexible, disordered loops
where host species changes, RBM need to adapt to better bind to new receptor variant
Inconsistency in the rate of mutations over different time periods and distances.
Host species and tissue tropism changes aren’t reflected in S/N ratio. Most pairs have an unrealistically high proportion of synonymous mutations even when the species hasn’t changed. The range of normality in other bat viral families is between 0.7 to 5.
These sequences should not be automatically accepted as evidence for SARS-CoV-2’s natural origin, or that unusually features such as the human-adapted RBM and NTD are natural. We need to have genuinely independent confirmation of the existence of similar viruses in nature and that the extraordinary evolutionary dynamics implied are valid.